
Hello, dear reader, and welcome to the first AIchemy Software Engineering Blog post. As you may know, AIchemy is the UK’s new AI for chemistry hub, tasked with developing and supporting the use of AI in chemistry research. As you may imagine, in order to effectively use AI in chemistry, a couple of requirements have to be met. Namely, we need an awful lot of data, and we need a way to make this data easily available to researchers. Fortunately, this is exactly the sort of thing the software engineering team (currently of size 1) is here to support.
Before looking into what exactly we did, it may be worth giving a brief overview of the data management practices currently at Imperial College, where I am currently based, and which are fairly representative of other universities in the UK. Experimental researchers use a variety of independent, college or department owned, facilities to collect experimental data. For example, say I synthesised a new compound and would like to determine what the molecular structure inside of my solution really looks like. Well, I will take my sample to the NMR facility, and take an NMR spectrum and I will take my sample to the mass spectrometry facility and collect a mass spectrum. The facilities provide me with the machines and training to use the machines, and will give me the output data of these machines. When it comes to publishing my results I have to ensure I kept all my data in a safe place, correctly labelled, and in a format that can be read by other people (though often not without substantial effort on their part).
This is essentially the state-of-play now, so let’s examine some of the issues researchers are starting to run into to motivate some of our work at AIchemy. Firstly, this workflow delegates data management to researchers. This may work on a small scale but data management is not an easy task. If you want to do your own data management, here’s just a small number of questions you may need to consider, off the top of my head:
- Which database should you use? SQL? NoSQL? PostgreSQL? MySQL? SQLite?
- What will your schema look like?
- How are data backups handled?
- Where should data be stored? Locally? On the cloud? How will this be paid for?
- How will data be accessed? SQL? A Python library? A web app?
- How will data be shared with others? Will this depend on external vs internal collaborators?
- Will changes to the data have an audit trail?
- How will schema changes be handled?
And so on . . . These questions are not just numerous, they also do not have obvious answers, and choices come with trade-offs. In an age where where FAIR (that is Findable, Accessible, Interoperable, Reusable) data principles are gaining traction, answers to these questions become increasingly important.
Sometimes, people are advised to gain additional training to help them answer these questions, however, I think it’s more important to ask why a person interested in chemical research should be considering these questions at all. The fact is delegating data management to individual research groups means that good data management practices will likely be inconsistently applied, less time can be spent on research, the same work is repeated over and over across different research groups and smaller groups are disadvantaged as they do not have the resources to build and maintain their own data management solution.
At AIchemy, our aim to ensure that research data is always FAIR, from the moment it is collected up to the point that it is published. Consider if instead when you visit a facility to collect your data, instead of being given data straight from the machine, you are given access to a database which not only gives you the data from the machine, but also metadata, such as how the data was collected, which processes were used for collection, who collected it, which chemicals were involved, etc. and all in a FAIR way. Finally the facility can allow you to publish your data to a DOI in the click of a button. Importantly for us at AIchemy, having data organized in this way removes much of the overhead from applying it in machine learning, removing much of the barrier to entry. As more people use this system, datasets which can be used for machine learning will grow larger and the performance of models can be expected to improve.
Work at the NMR Facility
With the preamble out of the way, let’s look at some of the work we have done at the Imperial College NMR facility, in conjunction with the two facility managers Stuart Elliot and Pete Haycock. We chose to start with the NMR facility because it is one of the most widely used facilities for characterizing compounds and so should be familiar to most researchers. It also has the capacity to perform a large number of NMR experiments, which means that if we manage to on-board the facility to a more robust data management system, we can quickly build up large datasets which can be used for machine learning.
The NMR facility has 4 main Bruker instruments, each of which is connected to a computer. The computers on each machine are disconnected from both the College network and from the general internet. When data is collected, it is transferred to a shared drive, which is accessible on the college network, and users can copy the data onto their own computers and manage it how they like. The data provided to the users has no metadata, apart from being stored in a folder shared by their research group.
Before we started looking at improvements to the data management system we ran a focus group with the current users of the facility and asked them what their main points of friction were. The main points were:
- The need to manually link NMR spectra across time, user, group, experiment, chemicals involved.
- Inefficiency with regard to experiment submission and monitoring. Submitting many experiments at once is a chore.
- Lack of information about queue times.
After some research into existing data management systems available for NMR data we ran into NOMAD NMR (https://www.nomad-nmr.uk/) developed by Tomas Lebl at the University of St Andrews. NOMAD NMR handles the submission and monitoring of NMR experiments along with collection of metadata and includes integrations with NMRium (https://www.nmrium.org/) for analysis of collected data. NMR allows you to download the raw data from the machines if you so wish, so you can continue to use your favorite NMR analysis software. NMR NOMAD also streamlines much of the day-to-day running of the NMR facility providing tools for user on-boarding, cost management and so on. Experiments collected with NOMAD can be tagged with metadata including the user, group, experiment, chemicals involved, and time taken, and allows you to search by these criteria. Experiments can also be grouped, for example if you have a set of experiments you ran for a specific research project.
NOMAD NMR provides a centralised database for all experiments, which can be accessed from anywhere on the college network. Data access can be be finely managed, which means data can be shared with other users or groups easily. Finally, and importantly for AIchemy, NOMAD NMR is built on a modern, well-established software stack, meaning it is easy to contribute to if we need to add features for our use case, and it has well understood performance characteristics as well as being easy to pick up and deploy in production.
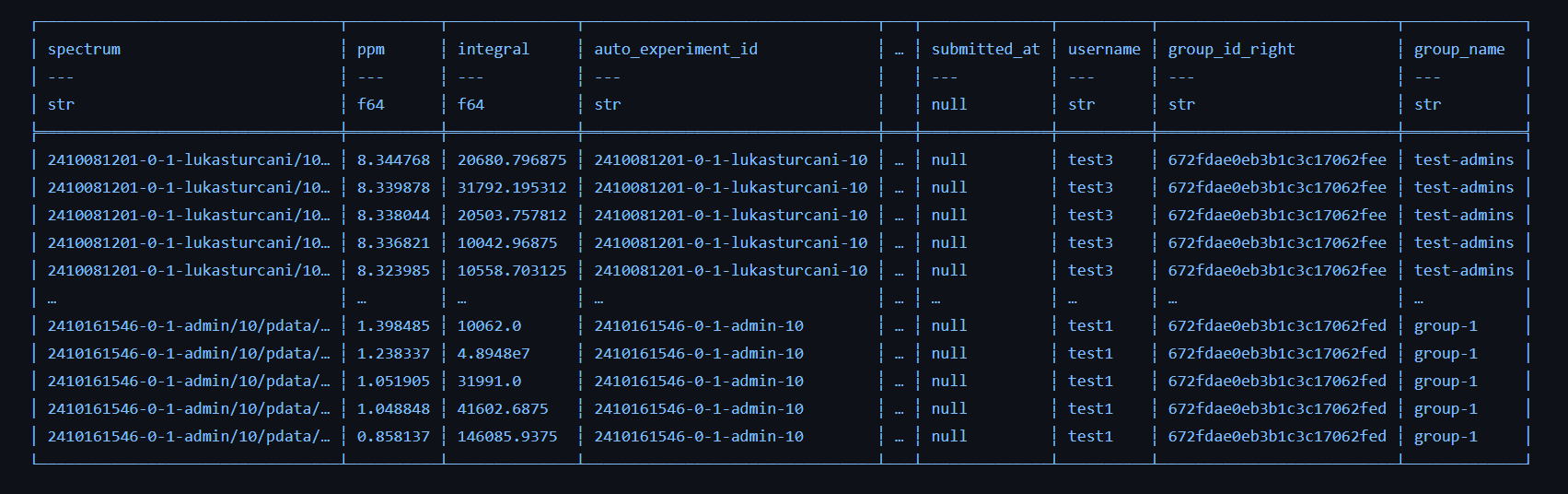
Out of the box, NOMAD NMR provided a much streamed lined experience for users and facility managers, however, it was intended to be used through a web interface. Our goal at AIchemy is to make it easy for researchers to machine learning in their workflows. For this, they need a programmatic interface, ideally in Python, so that they can easily get all of their NMR data in a format they can feed into their machine learning framework of choice. This is was not something NOMAD NMR provided out of the box, but fortunately, because NOMAD NMR is built with standard web technologies, it is easy to extend and integrate with other tools. At AIchemy, we built a Python library aitomic (https://aitomic.readthedocs.io). The library allows you to pull data from NOMAD NMR in just a couple lines of code. The data retrieved from NOMAD is available as data frames and already includes all the peaks in the spectra as well as their integrals. Lets see it in action:
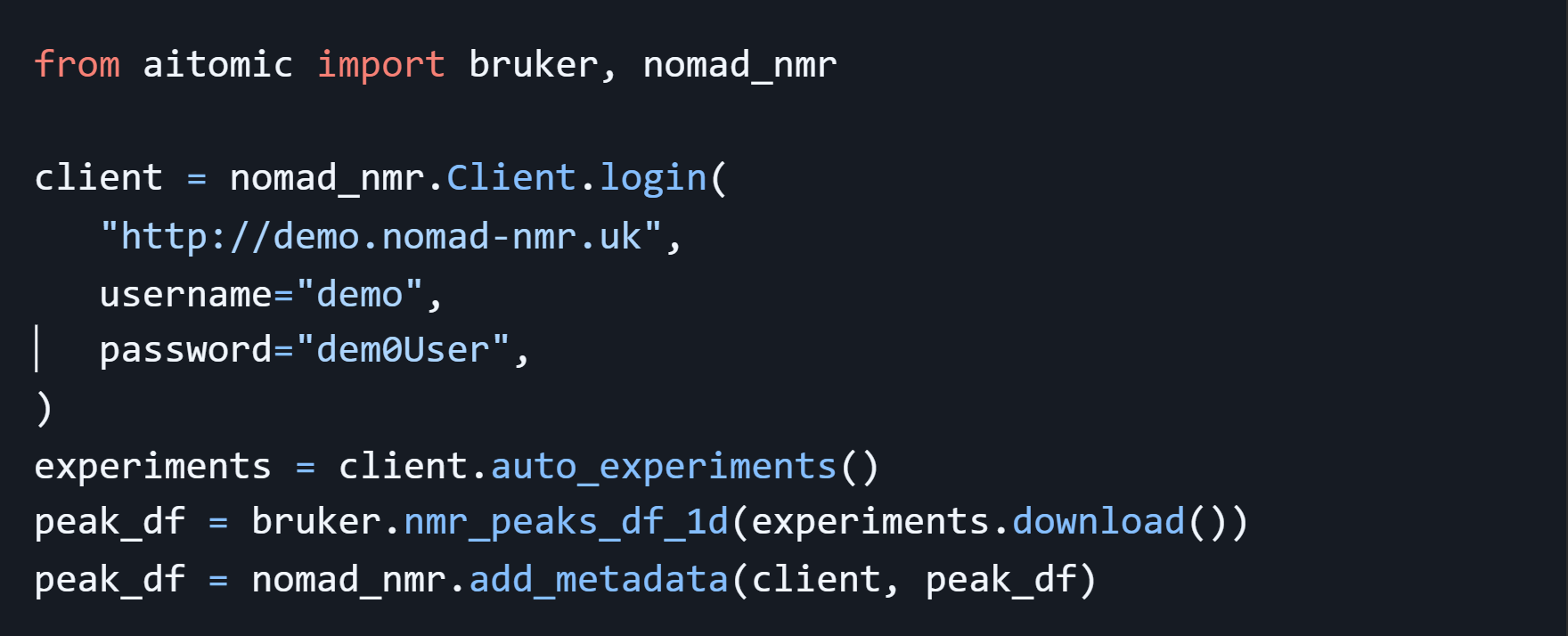
Our data frame looks something like this:
aitomic also allows you to download your raw data from the NOMAD server among other things. You can get it easily with pip:
pip install aitomic
aitomic comes with a fair number of examples, so make sure you check out the docs.
What’s next for AIchemy?
In this post, we took you through some of the initial work we have done at AIchemy. Going forward we hope to work with more facilities to on-board them onto good data management practices and provide toolkits to help them easily manage their data and use it for their research. We will continue to make updates to aitomic as we work with more facilities and data providers.