
Alchemy’s five forerunner projects are the initial research focus and cover a variety of different AI for Chemistry topics and challenges, applicable to both industry and academia, with partners ‘mapping’ onto projects as they develop. Future challenges facing the industrial chemical sector will be identified via our Industry Partners and translated to live research projects, facilitated through Alchemy’s funding calls.
>_F1: Human-in-the-Loop
>_F2: Large-Scale Crystal Structure Prediction
>_F3: Data-Driven Materials Discovery
>_F4: Generative AI for Small Molecules and Materials
>_F5: Identifying and Optimising Reaction Mechanisms
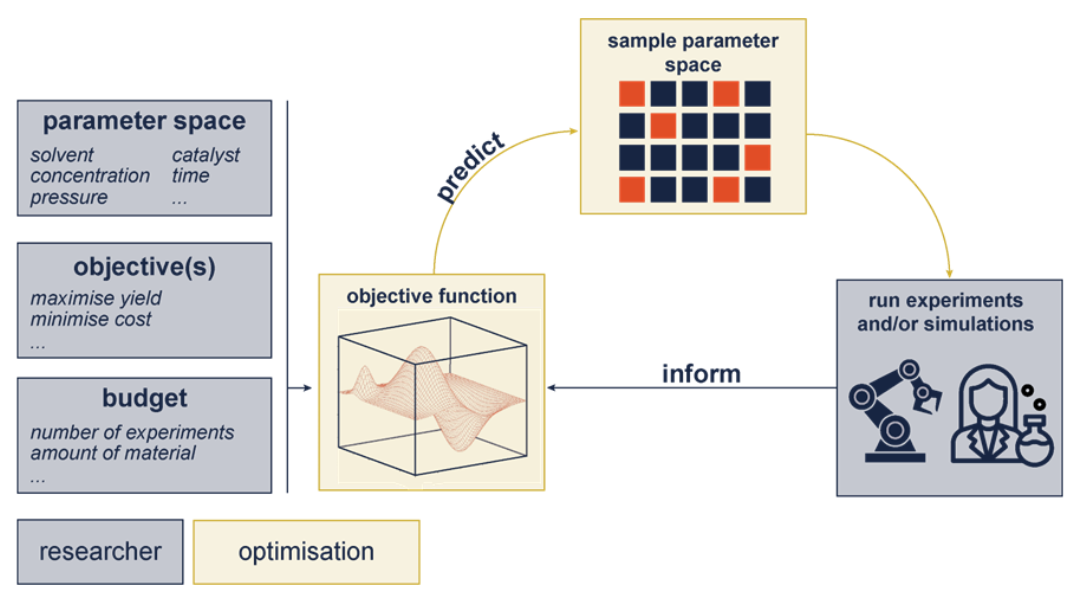
Combining human hypothesis and Bayesian Optimisation will allow chemists to build human intelligence and chemical knowledge into closed-loop AI-driven robotic experiments. The synergy between a human in the loop approach and robotics will enable the accelerated discovery of materials and properties.

University of Liverpool
Prof. Andy Cooper, Dr Xenofon Evangelopoulos
Research Update 1
Scientists from the Departments of Computer Science and Chemistry at the University of Liverpool have developed BORA – a “research assistant” powered by AI that helps accelerate scientific discovery. By combining the reasoning abilities of large language models (LLMs) with Bayesian optimisation, BORA can sift through complex, high-dimensional problems that usually take months of trial and error. Instead of getting stuck or wasting time, it uses literature-guided insights to suggest smarter experiments, explain its choices, and adapt on the fly. Tested on real challenges from solar energy to crop yields and new materials, BORA consistently found better solutions faster. This breakthrough shows how AI can act as a creative partner in science – not just crunching numbers but helping researchers explore uncharted territory with speed and confidence. The work of BORA has been published in the International Joint Conference of Artificial Intelligence (IJCAI), 2025 in Montreal.
Read more: https://ijcai-preprints.s3.us-west-1.amazonaws.com/2025/4998.pdf.
Research Update 2
In a follow-up study on Liverpool’s AI-powered “research assistant” for speeding up scientific discovery – BORA – the team from the Department of Chemistry carried out a detailed series of ablation studies to understand exactly what makes BORA effective, testing different large language models, optimisation settings, prompting strategies and batch sizes. Crucially, they also asked whether commercial large language models could outperform BORA on their own. The results showed that combining LLM reasoning with Bayesian optimisation remained a powerful and reliable approach, consistently outperforming Bayesian optimisation alone and often delivering stronger, more robust performance than LLM-only methods. The study also revealed when standalone LLMs can work well, helping to clarify the strengths, limits and future potential of AI-guided closed-loop experimentation. This work highlights the importance of rigorous benchmarking in autonomous science and shows how hybrid AI systems such as BORA can provide both insight and practical advantage in complex discovery problems. The work has been published in Digital Discovery, 2026.
The prediction of material structures and properties, particularly organic materials, remains an important challenge to chemists. Here the focus is to address the limited availability of data for training ‘inverse design’ methods for organic materials and to provide a step change in the quantity of data available to describe relationships between molecular structure, crystal structure, and functional properties.
University of Southampton
Prof. Graeme Day, Prof. Jeremy Frey
Research Update 1
AIchemy researchers based at the University of Southampton have created the largest dataset of computed organic crystal structures to date, performing over one thousand large-scale crystal structure prediction (CSP) simulations. CSP enables chemists to explore enormous areas of chemical space and explore the full range of possible crystal packings for a molecule, providing insights beyond the structures observed experimentally. In this project, the team successfully reproduced over 99% of known crystal structures and correctly identified the most stable forms in the majority of cases. The resulting dataset of more than four million hypothetical structures is openly available and has already been used to train proof-of-concept machine learning models. These include a neural-network correction that improves the accuracy of energy rankings and a message-passing neural network that can re-optimise predicted structures to better match experimental data. This work demonstrates the predictive power of CSP methods when applied at scale and establishes a valuable resource for advancing computational materials discovery. The study has been reported in Faraday Discussions (DOI: 10.1039/d4fd00105b) and provides a foundation for new applications of artificial intelligence in the design and understanding of molecular crystals.
Read this study here: https://pubs.rsc.org/en/content/articlelanding/2025/fd/d4fd00105b
Research Update 2
Building on the large scale crystal structure prediction database published last year, the dataset has been extended to include molecules with conformational flexibility by adding molecules with hydroxyl, amine and carboxylic acid functional groups. The current database includes predicted crystal structure landscapes for over 1,600 molecules, which includes over 40 million unique, lattice energy minimised crystal structures. As a demonstration of the value of this dataset, density functional theory calculations were performed on 2.2 million crystal structures from the dataset, from which a series of transferable machine learned potentials, MACE-CSP, have been trained. The accuracy of these models has been demonstrated in crystal structure prediction tests, reproducing known crystal structures accurately and, in most cases at the global energy minimum (58% of cases) or within 2 kJ/mol of the global minimum (87%).
Read this study here: https://chemrxiv.org/doi/full/10.26434/chemrxiv.15000584/v1
The literature available describing research undertaken on material classes and functions is vast and at times under utilised. This work aims to build and execute a ‘design-to-device’ pipeline for materials discovery for a chemical application via: AI-driven data sourcing, machine-learning predictions and high throughput experimental validation.
University of Cambridge
Prof. Jacqui Cole
Research Update 1
The AIchemy team at the University of Cambridge have developed machine learning models that can automatically determine molecular substructures directly from experimental nuclear magnetic resonance (NMR) spectra. Traditionally, interpreting NMR spectra requires expert analysis, which is time consuming and prone to human error. By training neural networks on more than 34,000 carbon-13 (¹³C) and 17,000 proton (¹H) spectra, the team showed that computational models can accurately correlate spectral features with molecular structure. They explored several architectures, including a multilayer perceptron combined with long short-term memory (MLP+LSTM) and convolutional neural networks (CNNs). The best-performing models achieved up to 88% accuracy for ¹³C NMR data when experimental metadata such as field strength, temperature, and solvent were included. CNN models offered a practical balance, providing high predictive accuracy while requiring only one-third of the computation time of LSTM-based models. Case studies on compounds such as aspirin, caffeine, and beta-sitosterol demonstrated that the models can correctly assign most substructures from spectra alone. This work highlights the potential of AI to accelerate chemical analysis and supports the development of automated workflows in spectroscopy and materials discovery.
Explore this work further here: https://pubs.acs.org/doi/10.1021/acs.jcim.5c00499
Chemical discovery through generative AI can identify novel molecule and materials classes with tailored compositions, structures and functions in areas of societal need. This will be achieved by using scalable generative models that harvest comprehensive chemical knowledge from large-scale multi-modal chemistry data to learn representations with desirable chemistry properties.

Imperial College London
Prof. Kim Jelfs, Dr. Alex Ganose
Research Update 1
Researchers from the Departments of Chemistry and Computer Science at Imperial College London are developing machine learning models to accelerate molecular structure elucidation from spectroscopy, with applications in drug discovery, metabolomics, and materials science.
Their approach builds on recent advances in fragment-based, spectra-conditioned generative modelling for molecular structures, and introduces two key extensions:
(1) scaling up to a large chemical space using a recently proposed multimodal spectroscopic dataset of ~790k molecule-spectra pairs, including compounds with up to 35 heavy atoms and 9 distinct chemical species.
(2) designing a hierarchical, multi-spectral encoder that combines information from IR and NMR input spectra.
In addition, they are benchmarking transformer baselines that directly translate raw spectra into molecular representations (SMILES/SELFIES), providing systematic comparisons across different methodologies. Looking ahead, our system can be fine-tuned on experimental datasets of molecule-spectra pairs as they become available and has the potential to be deployed as a web interface where users can upload spectra and obtain candidate molecular structures.
Research Update 2
Recently, NMIRacle was accepted at the ICLR 2026 Workshop on AI for Accelerated Materials Design. In this work, we investigate leveraging IR and NMR spectra for automated molecular characterization. NMIRacle leverages transformer-based architectures for associating spectral features to molecular functional groups, enabling end-to-end molecular structure elucidation under minimal input assumptions.
In parallel, I have been developing a hybrid autoregressive-diffusion framework for 3D molecular generation. The approach combines a causal transformer operating over latent representations with a diffusion-based, next-token generator, enabling sequential construction of molecules while retaining high-quality geometric fidelity.
Together, these projects reflect a broader goal of integrating experimental observables with generative modelling, to improve both molecular understanding and design.
The ability and speed at which we are able predict new materials is continuously increasing. This identifies the need for new methods to accelerate the rate at which materials can move from ‘prediction’ to reality.
Here, we set out to develop an automated platform for reaction mechanism discovery employing multifidelity ML models that combine computational and spectroscopic data.
Imperial College London
Dr. Alex Ganose, Dr. Becky Greenaway, Prof. Kim Jelfs, Prof. Ruth Misener
Research Update 1
Researchers from the Departments of Chemistry and Computer Science at Imperial College London are leveraging multi-fidelity AI to accelerate the discovery of new catalytic systems and the understanding of reaction mechanisms. The team have developed a tailored data-driven pipeline focused on porous materials for catalysis, integrating high-throughput screening with reaction data curation, transfer learning and Bayesian optimisation. By combining insights from in-house experiments with diverse external datasets, the researchers are optimising data-efficient AI models to identify improved conditions and emerging design principles for next-generation catalytic materials.
Research Update 2
The Forerunner 5 project has reached an exciting turning point, moving from framework development and testing to active, autonomous discovery. We have successfully launched our first iterations of extensive screening, allowing us to efficiently navigate the vast chemical space of our target reactions.
To meet the specific demands of our research, we have significantly updated our internal screening code. This custom enhancement enables our models to handle the unique, complex constraints inherent to the investigated materials in the most accurate and efficient way.
This enables us to drastically reduce discovery timelines and establish a new design approach for high-performance catalytic systems